
Hyaline Fibromatosis Syndrome (HFS) is a rare but severe genetic disease that affects babies, children, and adults. Hyaline, a glassy substance, accumulates in the skin and various organs, and causes painful deformities that can lead to an early death.
HFS is caused by mutation of a gene called Capillary Morphogenesis Gene 2 or CMG2. It is also abbreviated as ANTXR2 because it makes cells sensitive to anthrax infections.
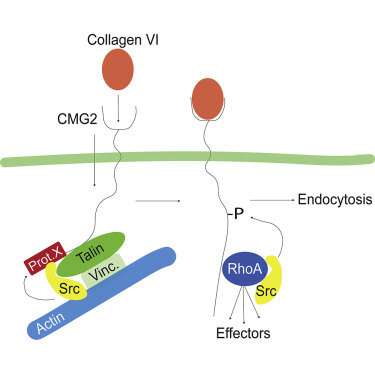
In 2017, the lab of Gisou van der Goot at EPFL showed that the CMG2 protein interacts with collagen VI, regulating its concentration inside the cell. In HFS, a mutation of the CMG2 gene prevents its protein to function, leading to accumulation of collagen VI in organs.
Now, the van der Goot lab and their colleagues have followed up on the work by showing that CMG2 interacts with specific components of the cell’s cytoskeleton. Publishing in Developmental Cell, the study reveals an entire network of such interactions.
Carefully “dissecting” the networks connected to CMG2, the researchers found that it binds Talin, a protein that helps connecting the key components of the cell’s skeleton, integrin and actin. In this way, Talin acts as the door through which CMG2 can interact with the cytoskeleton—but only when the latter is free from any ligand.
The paper describes the process by which CMG2 releases talin “picks up” a cytoskeleton regulator called RhoA, as well as its effector proteins. The scientists identify the specific parts of CMG2 that are responsible for these interactions.
Looking at human fibroblasts, the scientists finally concluded that the way CMG2 contributes to the degradation of excessive collagen VI is by switching between Talin and RhoA. The authors write: “This explains why HFS mutations in the cytoskeleton binding domain leads to dysregulation of extracellular matrix homeostasis.”
Source: Read Full Article