First described in 2020 by researchers from the US National Institutes of Health (NIH), VEXAS syndrome is a systemic autoinflammatory disease of undefined origin. Its name is an acronym: Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic. The prevalence of this syndrome is unknown, but it is not so rare. As it is an X-linked disease, men are predominantly affected.
First Identification
The NIH team screened the exomes and genomes of 2560 individuals. Of this group, 1477 had been referred because of undiagnosed recurrent fevers, systemic inflammation, or both, and 1083 were affected by atypical, unclassified disorders. The researchers identified 25 men with a somatic mutation in the ubiquitin-like modifier activating enzyme 1 (UBA1) gene, which is involved in the protein ubiquitylation system. This posttranslational modification has a pleiotropic function that likely explains the clinical heterogeneity seen in VEXAS patients: regulation of protein turnover, especially those involved in the cell cycle, cell death, and signal transduction. Ubiquitylation is also involved in nonproteolytic functions, such as assembly of multiprotein complexes, intracellular signaling, inflammatory signaling, and DNA repair.
Clinical Presentation
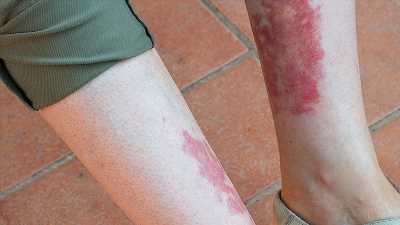
The clinicobiological presentation of VEXAS syndrome is very heterogeneous. Typically, patients present with a systemic inflammatory disease with unexplained episodes of fever, involvement of the lungs, skin, blood vessels, and joints. Molecular diagnosis is made by the sequencing of UBA1.
Most patients present with the characteristic clinical signs of other inflammatory diseases, such as polyarteritis nodosa and recurrent polychondritis. But VEXAS patients are at high risk of developing hematologic conditions. Indeed, the following were seen among the 25 participants in the NIH study: macrocytic anemia (96%), venous thromboembolism (44%), myelodysplastic syndrome (24%), and multiple myeloma or monoclonal gammopathy of undetermined significance (20%).
In VEXAS patients, levels of serum inflammatory markers are increased. These markers include tumor necrosis factor, interleukin-8, interleukin-6, interferon-inducible protein-10, interferon-gamma, C-reactive protein. In addition, there is aberrant activation of innate immune-signaling pathways.
In a large-scale analysis of a multicenter case series of 116 French patients, researchers found that VEXAS syndrome primarily affected men. The disease was progressive, and onset occurred after age 50 years. These patients can be divided into three phenotypically distinct clusters on the basis of integration of clinical and biological data. In the 58 cases in which myelodysplastic syndrome was present, the mortality rates were higher. The researchers also reported that the UBA1 p.Met41L mutation was associated with a better prognosis.
Treatment Data
VEXAS syndrome resists the classical therapeutic arsenal. Patients require high-dose glucocorticoids, and prognosis appears to be poor. The available treatment data are retrospective. Of the 25 participants in the NIH study, 40% died within 5 years from disease-related causes or complications related to treatment. Among the promising therapeutic avenues is the use of inhibitors of the Janus kinase pathway.
This article was translated from Univadis France.
Source: Read Full Article